
作者,Evil Genius
农村老家躺平中。。。。
关于疾病的研究,从基因组到转录组,到如今的空间组,每个组学都有其独特的角度,那么空间组研究的空间细胞结构关系对疾病的影响,是目前最缺失的一环。
之前在分享文章中,美洲人和非洲人虽然拥有同样的细胞类型,但是在癌症用药上缺有不同的治疗策略,就是因为不同人种细胞的空间排布不同。
今天我们分享文献

知识积累
慢性阻塞性肺疾病(COPD)在临床与分子层面均存在异质性。
整合单核RNA测序、高分辨率空间转录组学、肺组织蛋白质组学及血浆蛋白质组学。
患者队列与实验设计
分析策略与假设验证:研究假设经典细胞类型比例变化及疾病相关细胞状态的出现是COPD异质性的基础。通过量化各细胞谱系内的细胞及细胞状态比例,并将其与临床指标进行相关性分析(校正年龄、性别、吸烟状态和体重指数等混杂因素)。除肺气肿指标按肺叶评估并额外校正解剖位置外,其余分析均以个体为单位。
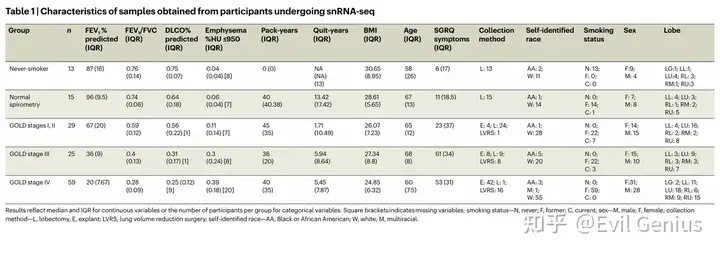
结果1、COPD中的炎症性非免疫细胞状态
非免疫谱系中识别出两类疾病相关细胞状态:炎症性非免疫细胞状态与修复/重塑细胞状态。
炎症性非免疫细胞状态广泛存在于内皮、上皮及成纤维细胞谱系中:
炎症内皮细胞(包括动脉型、普通毛细血管型、气体交换型):高表达炎症介质(NFKB1、IRF1、IL6等)并富集TNF、IFNγ、IL-1信号通路
炎症上皮细胞(AT2i、AT1i、分泌细胞i):特征基因包括AT2i中的SGPP2/CSF3/IL4R、AT1i中的IL32/IRF1及分泌细胞i中的CXCL趋化因子簇
炎症成纤维细胞(肺泡型、外膜型):表达NF-κB亚基、IRF1及与血管生成、组织重塑相关的因子(PLAUR、VEGF等)
关键发现:
传统认为特发性肺纤维化(IPF)相关的CTHRC1+成纤维细胞与ABCs在无影像学纤维化的COPD肺叶中被检测到
免疫荧光共染色显示它们在非成纤维灶的重塑区域共定位
贝勒队列中CTHRC1+成纤维细胞在疾病严重者中增多
ABCs在COPD中可能与CTHRC1+成纤维细胞协同参与跨疾病的共同病理过程
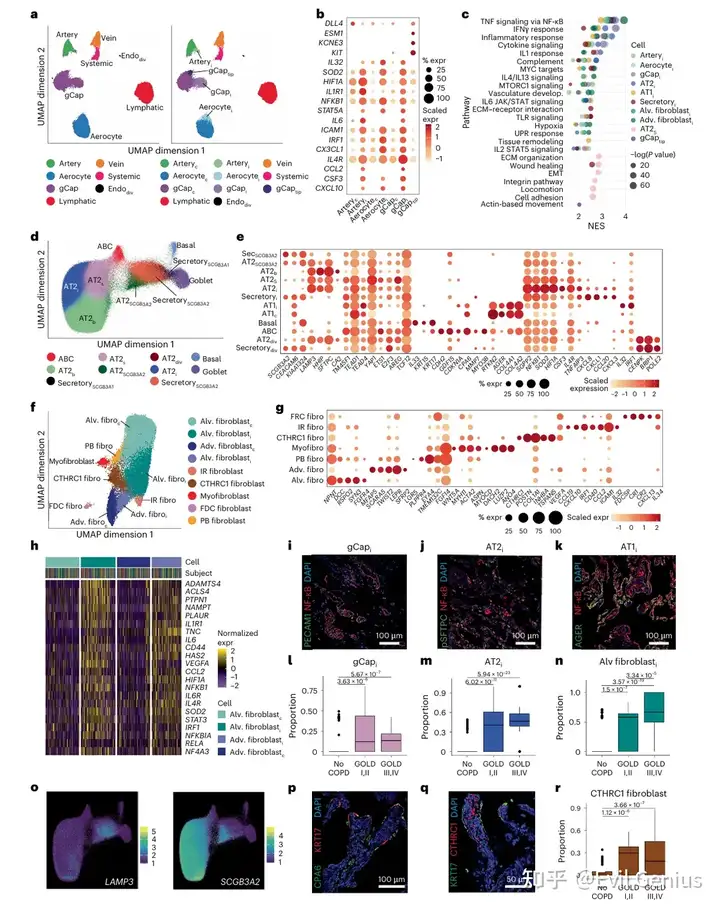
结果2、炎症、修复与纤维化细胞状态动态变化
炎症性非免疫细胞:
增加趋势:在COPD中增多,且随GOLD分期进展及肺叶特异性肺气肿加重而持续扩张
临床相关性:其比例与气流受限、一氧化碳弥散能力下降、肺气肿程度及症状评分呈正相关
个体异质性:在重症患者中比例差异显著,凸显个体间炎症反应的异质性
与吸烟无关:未发现与吸烟包年数或戒烟时长相关,提示戒烟后炎症持续存在可能存在其他驱动因素
修复性上皮细胞状态(包括AT2S、AT2SCGB3A2、AT2div、分泌细胞div):
吸烟诱导:在现/既往吸烟者中富集,表明其出现与损伤相关
动态变化:在COPD早期达到峰值,随疾病进展而减少
负向关联:AT2div与AT2SCGB3A2丰度与多项疾病严重程度指标呈负相关
修复性内皮细胞状态(Endodiv、gCaptip):
晚期减少:在疾病晚期减少,与疾病严重程度呈负相关
促纤维化细胞状态(ABCs与CTHRC1+成纤维细胞):
进行性扩张:随GOLD分期进展而增加,其比例与多项疾病指标正相关
病理转变:提示疾病进展过程中存在从早期修复向持续炎症及促纤维化反应的转化
其他成纤维细胞亚群:
区域性成纤维细胞减少:包括支气管周围成纤维细胞(LGR5+)和肌成纤维细胞(WNT5A+),其比例随GOLD分期及肺气肿加重而下降
潜在影响:这些区域性细胞群的减少可能削弱组织特异性修复能力,或直接反映晚期疾病的组织破坏
结论:COPD进展伴随细胞状态动态演变——早期修复反应逐渐被持续炎症与促纤维化过程取代,同时区域性修复细胞群丢失,共同推动疾病向晚期发展。
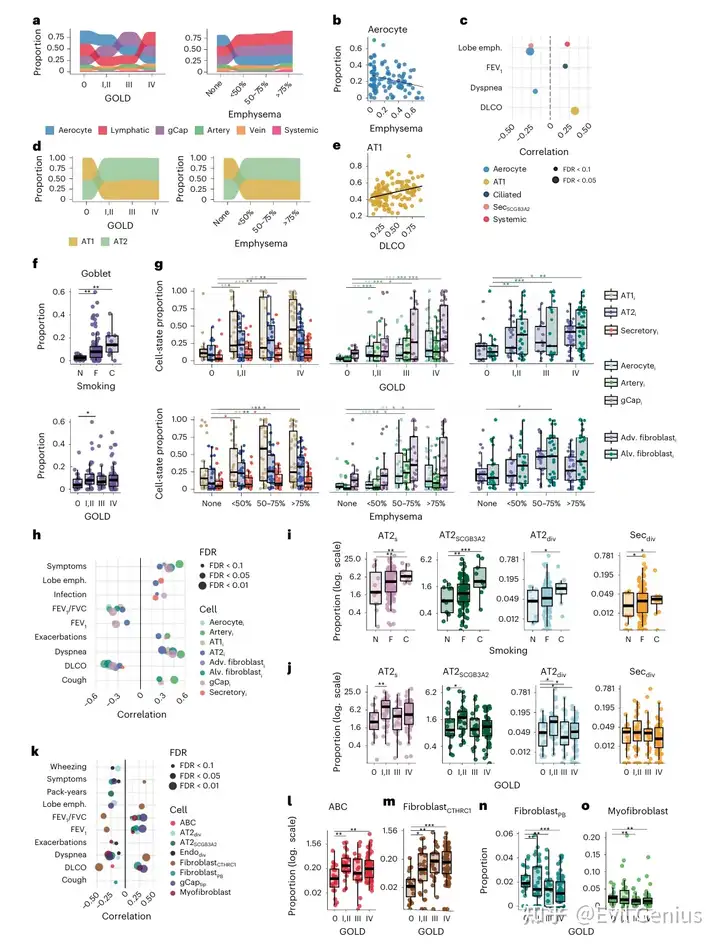
结果3、免疫细胞与淋巴成纤维细胞
巨噬细胞亚群分析:
识别出肺泡巨噬细胞、单核来源巨噬细胞、单核细胞及两种间质巨噬细胞(IM)亚群:
IM1:表达经典标志物(STAB1, F13A1)
IMCHIT1:高表达促纤维化基因(CHIT1, CHI3L1等),与严重气流阻塞、高肺气肿负荷、吸烟及其他临床指标关联最强
IM在COPD中整体增多,主要由促纤维化亚群IMCHIT1驱动,其富集程度与疾病严重程度正相关,并通过免疫荧光染色验证
炎症性肺泡巨噬细胞状态:
包括AMCSF1、AMNR4A与AMinflam,均显示细胞因子/趋化因子信号增强,分别与肺气肿、GOLD分期及急性加重不同程度相关
其他巨噬细胞群体:
AMp21:高表达DNA损伤应答基因(TP53, CDKN1A),在吸烟者及肺气肿程度<50%的患者中增多
AMHSP:富集热休克蛋白基因
AMIFIT:表达干扰素刺激基因
免疫细胞动态变化:
单核来源巨噬细胞在吸烟者及肺气肿程度<50%的肺组织中升高
增殖性巨噬细胞随疾病进展减少
B细胞与浆细胞在COPD多背景下增多
CD8+ T细胞与吸烟包年数正相关
肥大细胞与迁移性树突状细胞在晚期疾病中减少
淋巴相关成纤维细胞:
CXCL13+ 成纤维网状细胞:与淋巴结构相关,参与生发中心形成及B细胞招募/抗原呈递
免疫调节性成纤维细胞:高表达IRF1、CCL2、CXCL10等免疫调节因子,参与T细胞免疫调控
二者在晚期GOLD分期及影像学肺气肿受累程度高的组织中增多
结论: COPD进展伴随特定免疫细胞亚群的动态改变,其中促纤维化间质巨噬细胞(IMCHIT1)及淋巴相关成纤维细胞的扩张与疾病严重程度密切相关,提示免疫-间质互作在COPD病理演进中的关键作用。
结果4、基于复合表型的细胞组成分析
临床表型聚类:
整合LTRC队列的110个分类变量与24个连续变量(肺功能、影像学肺气肿、吸烟史、症状及功能状态),识别出7个临床聚类,代表不同COPD复合表型
各聚类对应明确的临床表型谱系:
聚类7:非吸烟者/肺功能正常
聚类1和6:轻度气流受限
聚类2和5:中度气流受限
聚类3和4:重度气流受限伴显著肺气肿(其中聚类2和4症状负担最重)
细胞组成变化:
下游分析合并聚类1+6、聚类2+5,形成5类平衡表型相似性与样本量的分组
细胞组成呈现方向性变化,与单指标分析趋势一致:
随疾病进展增加:CTHRC1+成纤维细胞、IMCHIT1巨噬细胞
随疾病进展减少:AT1细胞、气体交换型毛细血管细胞
关键发现:炎症性非免疫细胞特异性富集于同时具有重度气流受限与高症状负担的患者,而其他细胞群仅与生理损伤相关,与症状无关
疾病相关细胞状态的共现模式:
基于细胞类型比例相似性矩阵与谱聚类,识别出5个共现细胞群落:
混合炎症/纤维化群落:富集炎症与重塑细胞群
健康群落:疾病相关细胞状态比例低
杯状细胞/AMNR4A巨噬细胞富集群落
免疫富集群落:高表达CD4+、CD8+ T细胞、B细胞及IMCHIT1巨噬细胞
高炎症群落:高度富集炎症性非免疫细胞与炎性巨噬细胞
临床关联:
群落2肺功能最佳(DLCO、FEV1、FEV1/FVC最高)
群落1和5肺功能最差、症状评分最高,其中群落5的喘息与急性加重最显著
结论: COPD存在生物学定义的疾病程序,其细胞共现模式可独立于临床分类区分不同病理机制,这些程序具有重叠但非一致的临床表现,为疾病异质性提供了细胞层面的解释框架。
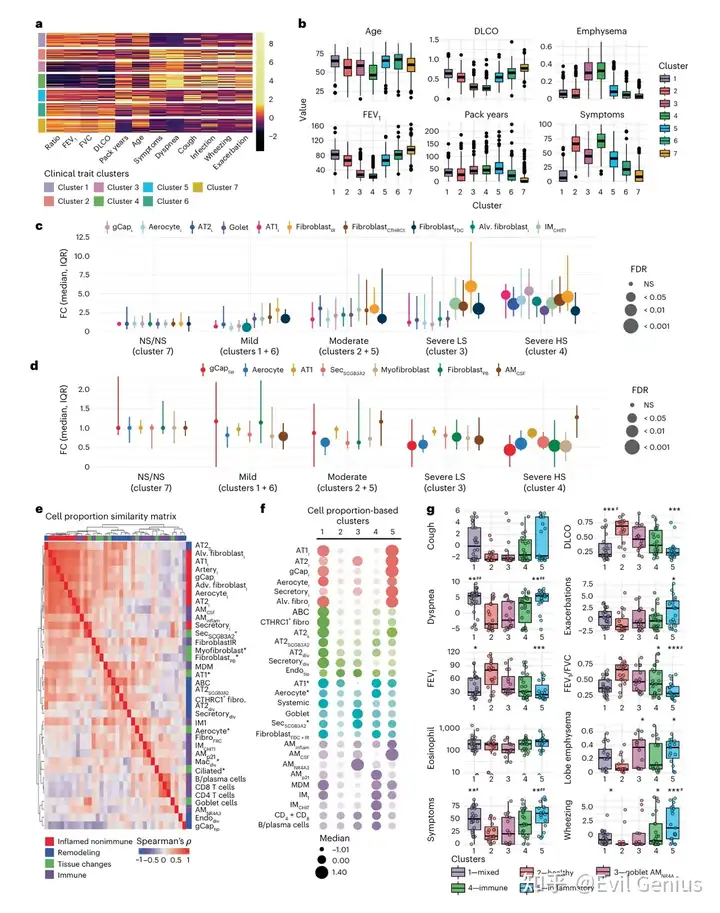
结果5、空间解析的细胞微环境
Xenium(480panel)
空间微环境界定:
基于细胞间欧氏距离构建k近邻图,依据物理共定位(非转录组相似性)定义空间微环境:
气道与大动脉微环境
低炎症非免疫细胞的肺实质微环境(LI-实质1和2)
再现snRNA-seq共现模式的微环境:
炎症非免疫细胞微环境
富含CTHRC1+成纤维细胞与ABCs的重塑微环境
肺泡与炎症巨噬细胞微环境
富含IMCHIT1巨噬细胞、T细胞与B细胞的免疫富集微环境
疾病关联与组织特征:
分布规律:低炎症实质微环境在非吸烟者与肺功能正常者中占主导,随COPD严重程度增加,炎症性、重塑性与免疫性微环境显著扩张
炎症微环境特征:组织学与转录叠加分析证实,炎症非免疫细胞常高表达炎症相关转录本,且常与富集IL1B、CXCL5等因子的巨噬细胞微环境相邻
重塑微环境特征:CTHRC1+成纤维细胞常与呈现细长形态、表达特征基因的ABCs相邻,二者多邻近富含B细胞及CD4/CD8/IMCHIT1的免疫微环境
COPD中的异常细胞在空间上形成具有明确组织定位的共现微环境,这些微环境的动态变化与疾病进展相关,为理解细胞间协同互作如何驱动组织层面病理提供了空间框架。
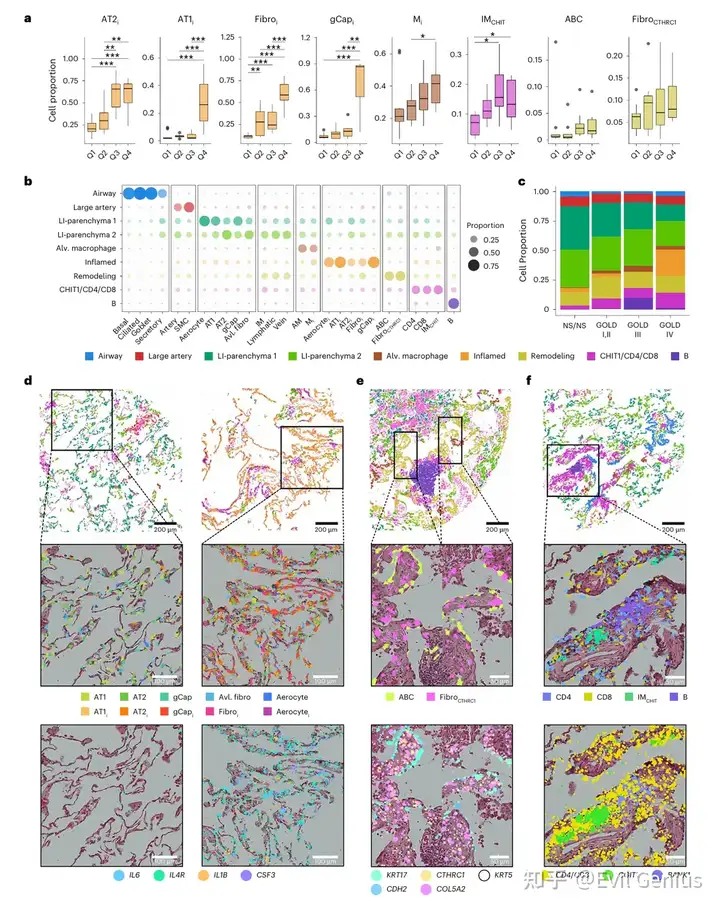
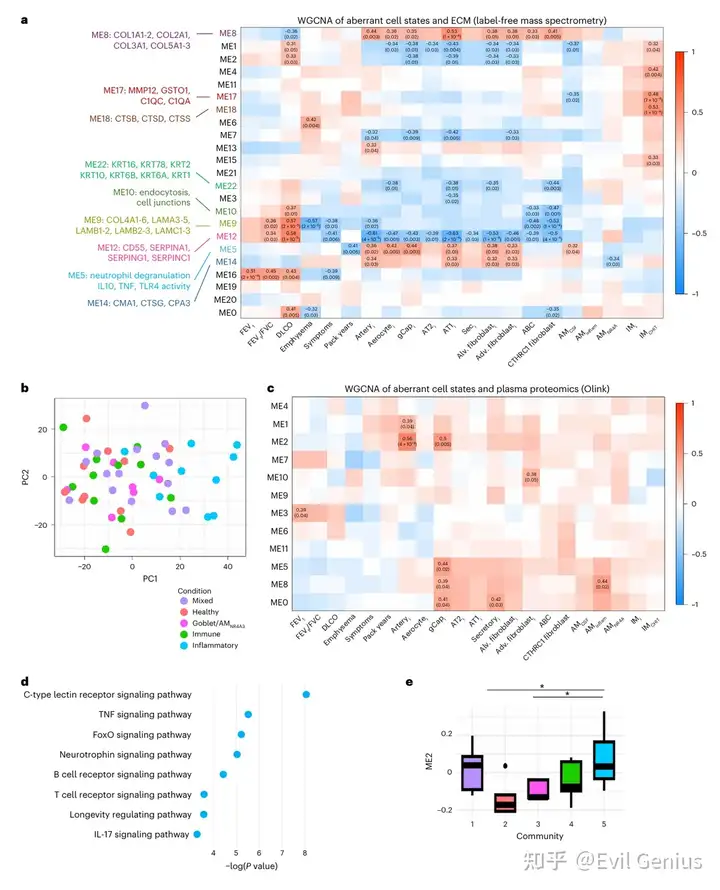
结果6、ECM蛋白质组学解析异常细胞状态与重塑的关联
ECM蛋白模块与细胞状态关联分析:
对匹配的脱细胞肺组织进行蛋白质组学分析,基于蛋白共现模式聚类为不同模块,并与异常细胞状态及临床表型进行关联
关键发现:
CTHRC1+成纤维细胞与炎症非免疫细胞与ME8模块(富含I、III、V型纤维状胶原)呈正相关,与ME22(角蛋白)及ME9(层粘连蛋白、IV型胶原)模块呈负相关
炎症非免疫细胞与ME5模块(富集炎症信号与中性粒细胞脱颗粒通路)正相关,与ME12模块(SERPINA1等补体/凝血抑制剂)负相关,提示抗蛋白酶活性降低可能加剧损伤与炎症
间质巨噬细胞(尤其IMCHIT1) 与富含COPD相关蛋白酶(MMP12、组织蛋白酶)的模块正相关,表明其可能是这些酶的主要来源
ECM蛋白组与疾病严重度的空间一致性:
ECM蛋白质组的主成分分析将样本独立分为与snRNA-seq相同的高严重度群落(群落1“混合炎症/纤维化”与群落5“高炎症”),证实这些微环境与ECM重塑蛋白的紧密关联
血浆蛋白质组学识别循环生物标志物:
对64例受试者的匹配血浆进行Olink Explore HT panel(5,420种蛋白标志物)检测,采用模块化分析
血浆模块ME2与炎症性动脉内皮细胞及普通毛细血管内皮细胞状态相关,富集炎症相关通路(C型凝集素受体、TNF、IL-17信号等)
ME2模块包含NF-κB调节因子、淋巴/髓系转录因子、细胞因子信号介质、天然免疫效应蛋白及凋亡相关蛋白
ME2特征评分在“高炎症”群落(群落5)中显著升高,提示其可能作为无创性生物标志物反映组织高炎症状态
异常细胞状态通过特定ECM蛋白模块驱动组织重塑,且其信号可延伸至循环系统,为COPD的病理机制解析及无创生物标志物开发提供了多组学依据。
结果7、COPD中的细胞自主性与细胞信号通路
细胞特异性基因表达模块,并评估其与临床特征的关系(校正性别、BMI、年龄和吸烟暴露)。
模块分析与中介效应:
与疾病中介或相关的模块富集于以下通路:炎症信号传导、生长与修复(WNT、TGF/BMP、生长因子)、衰老相关通路(端粒维持、DNA损伤应答、mTOR信号传导、自噬、应激反应),以及细胞迁移、细胞外基质相互作用、细胞死亡、细胞周期调控和蛋白质代谢。
细胞状态丰度的中介分析显示,IMCHIT1巨噬细胞介导了吸烟对呼吸困难、FEV1和FEV1/FVC的负面影响,而AT1细胞则对呼吸困难具有保护作用。
异常细胞状态的信号传导网络:
为阐明维持异常细胞状态的信号机制,我们对snRNA-seq数据进行了配体-受体互作推断,将异常细胞视为炎症/重塑信号的接收者和来源。
利用空间转录组数据(通过局部莫兰指数量化空间自相关性)验证这些相互作用,结果显示异常细胞中或疾病严重程度增加时,配体-受体共定位更为显著。
炎症非免疫细胞相较于非炎症状态同类细胞,接收更强的IL4R、IL33、IL6、IL-1、IFN和TNF信号传导。
IMCHIT1巨噬细胞相较于IM1巨噬细胞,接收更多SEMA3A、CSF1、IL4R和TNF信号。
作为信号产生细胞:
炎症内皮细胞是CXCL10、CXCL11和CSF3的主要来源;
炎症成纤维细胞(肺泡和网状)高表达CCL2、VEGF、IL15和IL33;
炎症巨噬细胞表达CXCL3、CXCL5、IL1B、IL1A和CCL2。
结论: 这些发现共同描绘了细胞类型特异性且空间组织化的信号网络,这些网络可能维持炎症或重塑微环境,并推动COPD疾病进展。

最后来看看xenium的分析方法

