
作者,Evil Genius
盗版商们,你们卖盗版无本万利赚了很多钱了,也该到用你们的时候了,pyrosetta和Gromacs的全系列中文教程,我觉得你们当仁不让。
多组学感觉已经来了,大家也要跟上点时代潮流了。
有之前的学员问如果喜欢科研,可以进课题组或者医院之类的,想法很好,但是不太现实,因为大家要明白,生存是第一位的,把做科研当成工作的,适合那种编制内,没有任何生存压力,安安稳稳做科研探索,我们普通人,农村出身,家里支撑不到这个地步。
无数的生信同事前辈都尝试走这个路,只有一位去了血研所的同事,稳定签了长期合同,工资足额发放,965工作制,完成科研分析工作即可,说白了其实还是商业制度,其他无一例外都失败了,没有类似编制的这种稳定因素,还是好好上班吧。如果以科研助理等形式进去了课题组,课题组老师把你当学生用,而你又没有稳定的身份,矛盾迟早爆发,到时候不仅坑了课题组,把自己也坑了。
当然,决定读这个课题组的博士,科研助理好好做一年是可以的。
做科研需要很高的精力,不断的探索,需要长期的稳定,像我这种有今天没明天的人,工作生活一团乱麻的底层人物,注定是要走商业模式的,科研模式走不下去。
今天我们分享知识,来源于

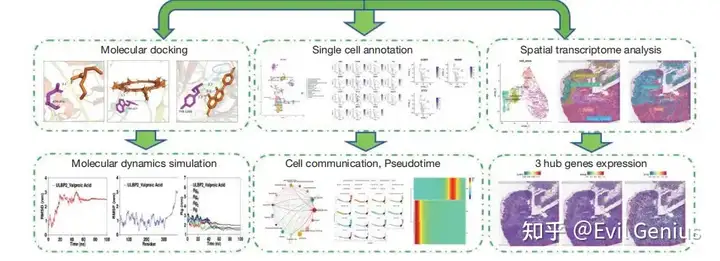
基因组 + 单细胞/空间 + 结构生物学,慢慢成为了一个整体,对人的要求也越来越高了,科研现在也要求完整的闭环,基因组 + 单细胞/空间在于发现科研的生物学问题,而如何解决这个问题(例如肿瘤疾病等),就需要分子对接/分子动力学,甚至还有体外实验,类器官、动物模式等。
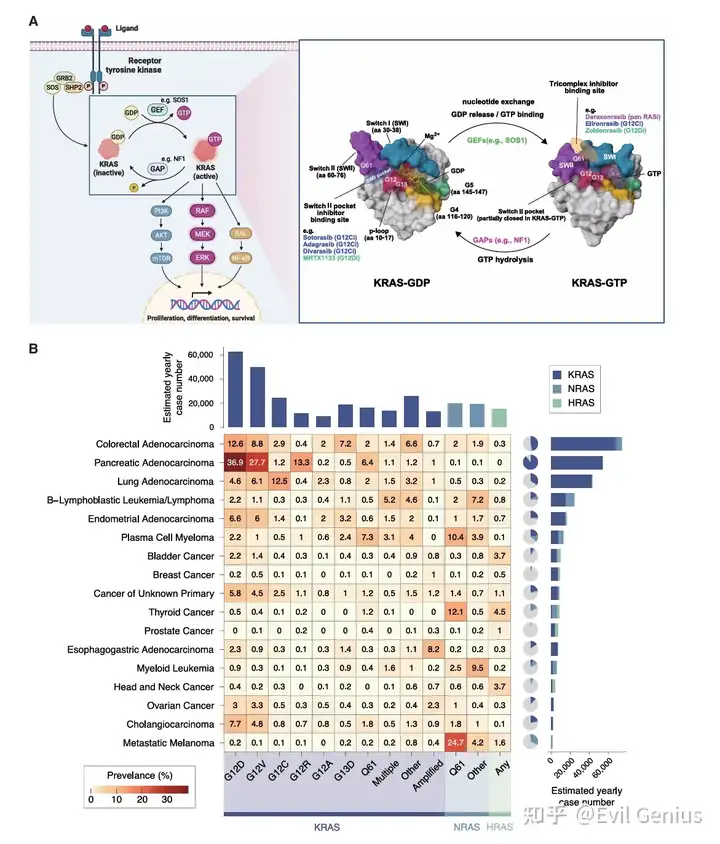
做WES,肿瘤研究的科研人员,RAS蛋白绝不陌生,RAS基因家族(KRAS、NRAS和HRAS)的突变约占人类癌症的20%,全球每年影响约340万新发患者。RAS蛋白在几乎所有细胞类型中普遍表达,其中KRAS是最主要的亚型。
体内研究表明,在基因工程小鼠模型中完全敲除KRAS基因会导致胚胎致死,而NRAS和HRAS对小鼠正常发育非必需,其功能可由KRAS代偿。然而,用HRAS替换KRAS可挽救KRAS敲除引起的胚胎致死效应,且在成年小鼠中敲除KRAS不会产生明显缺陷,这表明单个RAS亚型对正常细胞存活并非必需。
RAS蛋白的结构与功能
RAS蛋白由189个氨基酸(HRAS、NRAS和KRAS-4A)或188个氨基酸(KRAS-4B)组成,各亚型间具有高度保守的催化G结构域。除该保守域外,C端高度可变区决定了各亚型在膜结合与功能特异性上的差异。
功能上,RAS蛋白属于GTP酶家族,作为二元分子开关在失活的GDP结合“关闭”状态与激活的GTP结合“开启”状态间循环。此循环受鸟嘌呤核苷酸交换因子(GEFs)和GTP酶激活蛋白(GAPs)紧密调控:如SOS1等GEFs催化核苷酸从GDP结合态向GTP结合态转换,而包括NF1在内的GAPs则通过显著提升RAS内在GTP水解速率使其回归失活状态。
生理状态下,GEF介导的RAS激活主要由外部刺激触发,尤其是各类生长因子与胞外受体结合后启动的受体酪氨酸激酶(RTK)信号级联反应。酪氨酸磷酸酶SHP2通过去磷酸化抑制位点、招募GRB2-SOS1复合体至激活的RTKs以促进RAS活化,同时通过JAK-STAT、PI3K-AKT及免疫调节通路介导更广泛的信号传递。
RAS激活后,其开关Ⅰ域和开关Ⅱ域发生构象变化,从而能够与多个下游效应因子相互作用,其中最典型的是不同RAF蛋白激酶(如RAF1)。这引发磷酸化级联反应,最终激活经典的RAF/MEK/ERK通路。尽管近期研究提示RAS驱动的癌症主要依赖ERK信号,其他已确认的RAS效应因子还包括PI3K(连接RAS与PI3K/AKT/mTOR级联)以及促进RAS样蛋白活化的RalGDS。这些信号通路共同调控细胞生长、增殖、迁移、代谢和分化等关键细胞功能。
致癌突变及其对RAS信号通路的影响
当RAS发生致癌突变或基因扩增时,其精密调控的循环状态被破坏,导致下游信号通路异常激活与细胞生长失控。超过90%的RAS突变发生在第12、13和61位密码子,这些突变引起RAS蛋白催化G结构域的构象变化,从而限制GAP介导的GTP水解。其结果是GTP结合态与GDP结合态RAS的比例急剧上升,使平衡向激活的“开启”状态倾斜。值得注意的是,根据突变类型的不同,部分RAS突变体保留着不同程度的GAP非依赖性内在GTP水解活性,使其仍具备部分自失活能力,并能在“开启”与“关闭”状态间持续循环。在常见的KRAS突变体中,KRASG12C具有最高的内在GTP水解活性,这一关键特性推动了首个突变特异性KRASGG12C“关闭”状态抑制剂的研发。
药物开发
第一代KRASG12C抑制剂:Sotorasib(AMG 510)和Adagrasib(MRTX849) 是首批获批的KRASG12C抑制剂。
耐药机制:
肿瘤可通过多种途径对KRASG12C抑制剂产生耐药,包括:
获得性KRAS二次突变(如G12D/R/V,G13D,Y96C等)。
上游信号再激活(如RTK、SHP2激活)。
旁路信号激活(如MET扩增、PI3K/AKT/mTOR通路激活)。
组织学转化等。
新一代抑制剂与联合策略:
新一代KRASG12C抑制剂:如Divarasib(GDC-6036)、Glecirasib等,具有更高的效力和选择性,早期临床数据显示了更高的缓解率(ORR >50%)和更长的PFS(>12个月),正在开展头对头III期研究。
联合疗法(核心发展方向):
垂直联合:与上游(SHP2i、SOS1i)或下游(RAF、MEK、ERKi)靶点抑制剂联用,以更完全地抑制MAPK通路,预防或克服耐药。
免疫联合:与PD-1/L1抑制剂联用,旨在将“冷”肿瘤转化为“热”肿瘤,增强抗肿瘤免疫。早期数据显示在NSCLC一线治疗中前景可观(如Adagrasib + Pembrolizumab)。
其他通路联合:与靶向PI3K/AKT、CDK4/6、EGFR(在结直肠癌)等通路的药物联用。
通过开发更强效的新一代抑制剂和设计合理的联合治疗方案,治疗的有效性和持久性正在不断提升。未来的重点是将治疗益处拓展至非G12C的KRAS突变患者,并最终实现对RAS驱动癌症更有效、更持久的控制。
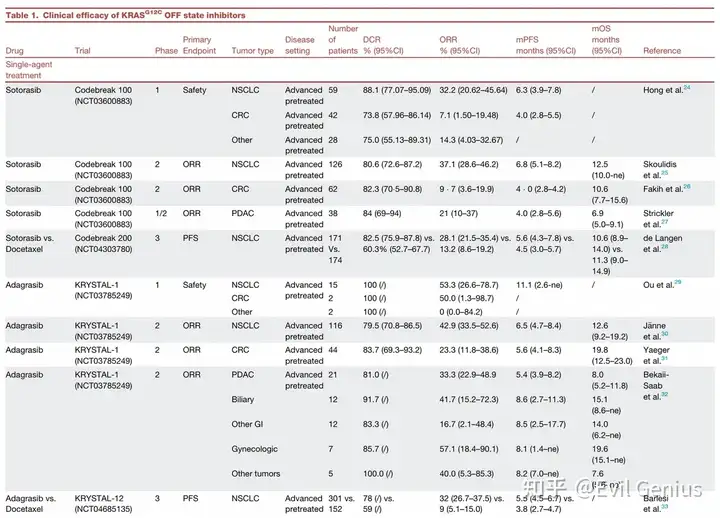
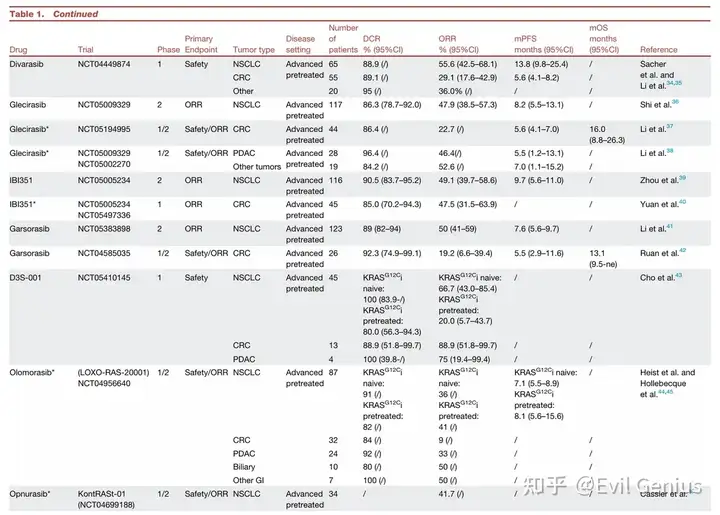
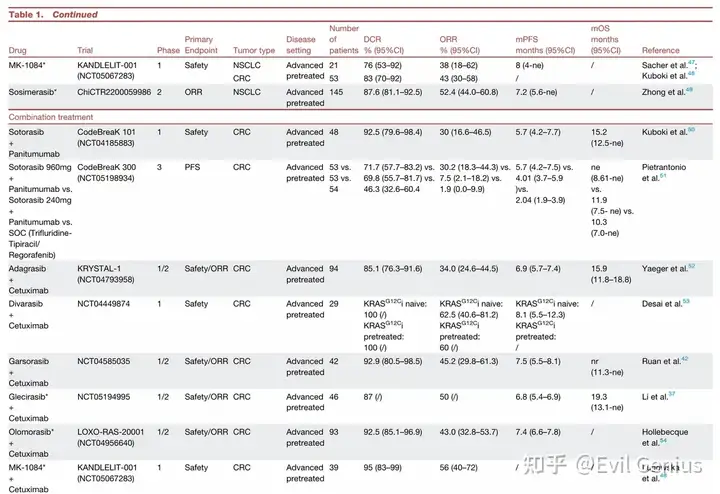
KRAS G12C 抑制剂临床经验总结
核心机制
这类小分子抑制剂通过结合并共价抑制处于失活、GDP结合状态的 KRAS G12C 突变蛋白发挥抗肿瘤作用。
非小细胞肺癌
疗效确切: KRAS G12C 是 NSCLC 中最常见的 KRAS 突变亚型。以索托拉西布和阿达格拉西布为代表的药物在经治的晚期 NSCLC 患者中显示出客观缓解率(ORR)约 32-43%,中位无进展生存期(PFS)约 6.3-6.5 个月,已获 FDA/EMA 有条件批准。
挑战与探索:
耐药性: 反应持续时间有限,存在原发性或获得性耐药。
联合治疗: 与免疫检查点抑制剂(如帕博利珠单抗)的早期联合方案显示出显著疗效(如阿达格拉西布联合治疗在 PD-L1 高表达患者中 ORR 达 61%),但也存在肝毒性等安全性挑战。多项 III 期联合治疗试验正在进行中。
新一代药物: 如迪瓦拉西布等新一代抑制剂显示出更高的体外效力和初步临床活性(ORR 达 55.6%),正在 III 期试验中与现有药物进行头对头比较。
结直肠癌中KRAS G12C抑制剂的应用总结
单药疗效有限
疗效数据:在转移性结直肠癌中,KRAS G12C“关闭”状态抑制剂的单药疗效显著低于非小细胞肺癌。
Sotorasib:客观缓解率仅9.7%,中位无进展生存期4.0个月。
其他药物(Adagrasib, Divarasib等):客观缓解率稍高(23%-29.1%),中位无进展生存期约5.6-7.8个月,但临床获益仍然短暂。
耐药机制:疗效受限的主要原因是EGFR介导的RAS/MAPK通路再激活。抑制KRAS G12C后,EGFR等受体酪氨酸激酶适应性上调,通过野生型RAS亚型激活旁路信号,导致快速耐药。
联合治疗成为突破关键
基于临床前模型的协同效应及BRAF V600E突变结直肠癌中垂直阻断RAS/MAPK通路的成功经验,KRAS G12C抑制剂联合抗EGFR抗体成为核心策略,并取得显著进展:
Adagrasib + 西妥昔单抗
在经治转移性结直肠癌患者中,客观缓解率提升至34%,中位无进展生存期6.9个月,中位总生存期15.9个月。
已获得FDA加速批准用于化疗难治性转移性结直肠癌。
确证性III期试验(KRYSTAL-10)已完成入组。
Divarasib + 西妥昔单抗
表现出迄今最高的客观缓解率(62.5%),且在既往接受过KRAS G12C抑制剂治疗的患者中仍有响应。
Sotorasib + 帕尼单抗
III期临床试验显示,与标准治疗相比,中位无进展生存期显著改善(5.6个月 vs 2.0个月),客观缓解率达30.2%,已获FDA批准。
一线治疗联合化疗(FOLFIRI)的初步数据显示客观缓解率高达78%,所有可评估患者均出现肿瘤缩小。
未来方向:向更前线治疗迈进
多项评估KRAS G12C抑制剂联合抗EGFR抗体及化疗用于晚期结直肠癌一线治疗的随机III期试验正在进行中。
这表明突变选择性KRAS G12C抑制剂有望很快被整合到结直肠癌的前线乃至(新)辅助治疗策略中。
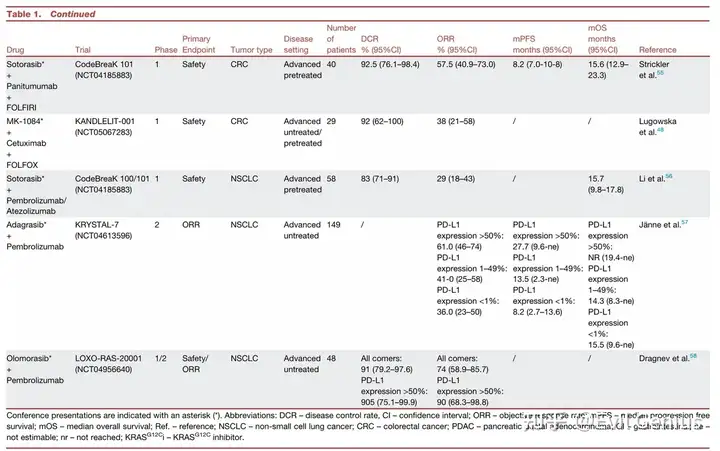
在其他恶性肿瘤中的应用前景
尽管目前批准适应症仅限于非小细胞肺癌和结直肠癌,但数据显示KRAS G12C抑制剂在其他瘤种中也具有潜在疗效,呈现“不限癌种”的治疗潜力:
胰腺导管腺癌:预后极差,治疗选择有限。Sotorasib和Adagrasib在经治患者中分别达到21%和33.3%的客观缓解率,中位无进展生存期约4-5.4个月。
其他肿瘤:在胆道癌、妇科恶性肿瘤等中也观察到鼓舞人心的初步疗效。
包括Divarasib、Glecirasib在内的新一代KRAS G12C抑制剂在这些瘤种中也报告了积极的初步数据。
胰腺导管腺癌及其他肿瘤
存在肿瘤无差别活性: 除了 NSCLC 和 CRC,KRAS G12C 抑制剂在 PDAC、胆道癌、妇科肿瘤等其他突变实体瘤中也显示出有前景的抗肿瘤活性(ORR 14.3% – 57.1%),为这类预后极差的肿瘤提供了新的治疗选择。
初步数据: 索托拉西布和阿达格拉西布在 PDAC 中的 ORR 分别为 21% 和 33.3%,新一代药物也报告了积极的初步疗效。
核心观察与未来方向
肿瘤类型差异: KRAS G12C 抑制剂的敏感性存在显著的肿瘤类型差异:NSCLC > PDAC > CRC(单药)。这提示不同肿瘤的适应性耐药机制和 KRAS 依赖性存在关键差异。
克服耐药: 针对 CRC 中 EGFR 介导的耐药,联合抗 EGFR 治疗已取得突破,使 ORR 提升至与 NSCLC 相当的水平。
未来策略: 研究的重点正转向多种联合策略以增强疗效和持久性,包括与 RAS/MAPK 通路上游(SHP2, SOS1 抑制剂)或下游(MEK, ERK 抑制剂)靶点、PI3K/AKT 通路抑制剂或 CDK4/6 抑制剂等进行联合治疗。
KRAS G12C 抑制剂耐药机制总结
原发性耐药
定义: 治疗前即存在、导致肿瘤对药物无反应的固有特征。
相关生物标志物:
共存RAS突变: 基线时存在其他RAS激活突变(如KRAS G12D)会导致无应答,但这种情况在初治肿瘤中罕见。
KEAP1等抑癌基因突变: 在NSCLC中,基线存在KEAP1、SMARCA4、CDKN2A突变(>25%的患者)与较差的临床结局相关。KEAP1失活可能通过激活NRF2促进耐药。但随机试验数据显示,KEAP1状态可能更多是预后而非预测性标志物,其通用性待验证。
ATM突变: CodeBreak 200试验中,ATM野生型患者从索托拉西布中获益更大,提示其可能是预测性生物标志物。
CRC/PDAC中的共存突变: TP53和PIK3CA突变在临床相关性上尚不明确。
获得性耐药:遗传机制
约60%的临床耐药患者可检测到获得性遗传改变,主要涉及RAS/MAPK通路。
检测方法: 循环肿瘤DNA测序是主要手段,但存在局限(如无法检测非遗传机制、肿瘤DNA脱落少等)。
核心原则: 耐药后肿瘤仍高度依赖RAS/MAPK信号,这为开发广谱KRAS抑制剂及垂直通路联合抑制提供了依据。
分类:
靶上改变: 直接影响KRAS蛋白本身(约占40%)。
KRAS扩增: 增加靶蛋白量,使药物“应接不暇”。
二次激活突变: 最常见于野生型等位基因(如G12D, G13D, Q61H),产生不受G12C抑制剂影响的新激活KRAS蛋白。广谱KRAS/RAS抑制剂可能克服此类耐药。
开关II口袋突变: 与G12C突变顺式发生(如R68, M72, H95, Y96),改变蛋白构象,阻止抑制剂结合。通过其他机制结合的下一代KRAS抑制剂可能有效。
靶外改变: 再激活下游或旁路信号。
上游调节因子: RTK(如EGFR)的扩增、融合或突变。
RAS旁系同源物: NRAS、HRAS的激活。
下游效应因子: BRAF、MAP2K1(MEK1)的激活事件最常见。
复杂性: 约1/4患者出现多个RAS/MAPK通路改变共存(在CRC中更常见),常为靶上和靶外改变同时出现,凸显了分子复杂性和针对多机制联合治疗的必要性。
获得性耐药:非遗传机制
约25%的患者在疾病进展时未检测到遗传驱动因素,表明非遗传机制至关重要。
研究挑战: 缺乏纵向肿瘤活检,难以在临床前模型中模拟。
主要类型:
适应性耐药: 最常见。KRASG12C抑制解除负反馈回路,导致多种RTK(如EGFR、HER2、FGFR、c-MET)快速激活,进而:
激活野生型RAS蛋白(NRAS/HRAS),绕过被抑制的KRASG12C。
将KRASG12C自身推向GTP结合的“激活状态”,使“失活状态”抑制剂无法有效结合。
CRC: EGFR是主导驱动因素,这解释了联合抗EGFR抗体疗效显著的原因。
其他肿瘤: RTK介导的耐药更异质,克服难度大。针对RTK的双/多功能抗体及SOS1、SHP2抑制剂是前景广阔的联合策略。
旁路通路激活:
YAP/TAZ/TEAD信号: KRASG12C抑制通过破坏细胞极性蛋白Scribble等功能,导致YAP入核,激活TEAD介导的转录(包括MRAS),进而通过SHOC2/PP1c复合物去磷酸化CRAF,最终再激活MAPK通路。临床前研究表明,联合TEAD抑制剂具有协同抗肿瘤潜力。
细胞可塑性:
上皮-间质转化: 由ZEB1、SNAIL等转录因子驱动,与对KRAS抑制剂等多种靶向药耐药相关。
组织学转化: 在NSCLC中观察到腺癌向鳞癌转化,或向肺泡/黏液分化状态转变,这反映了肿瘤通过转录重编程降低对KRAS信号的依赖性。
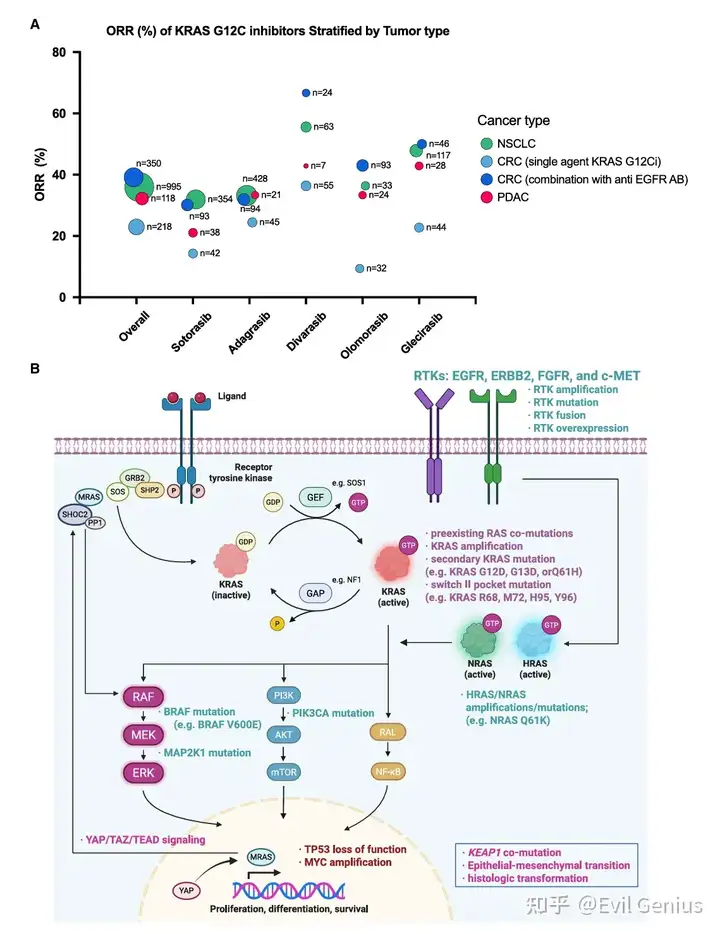
新型(K)RAS抑制剂发展格局总结
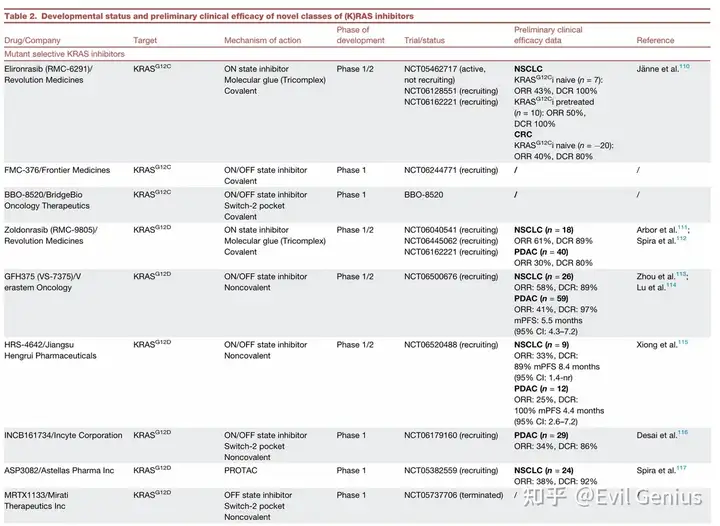
现有靶向开关II口袋的KRAS G12C失活状态抑制剂(如索托拉西布、阿达格拉西布)虽已显示临床疗效,但其治疗持久性因快速耐药而受限。同时,KRAS G12C突变仅占所有KRAS突变的约3%,绝大多数KRAS驱动肿瘤患者无法受益,这迫切需要开发针对更常见突变(如G12D、G12V)的抑制剂。
不同KRAS突变的生化特性与挑战
KRAS G12C: 保留GTP水解活性,可在GTP结合(激活态)与GDP结合(失活态)间循环,失活态相对可及,便于设计靶向半胱氨酸的共价抑制剂。
KRAS G12D: 突变为带负电、极性、侧链较小的天冬氨酸,显著降低GTP水解活性,使蛋白主要锁在激活态。缺乏可共价结合的半胱氨酸,静电环境和持续激活状态使其成为传统失活态抑制剂的挑战性靶点,需要激活态抑制剂等新策略。
KRAS G12V: 突变为庞大、疏水的缬氨酸,产生显著空间位阻和构象刚性,进一步抑制GTP水解,偏向激活态。缺乏反应性半胱氨酸或其他合适官能团,对抑制剂设计构成重大挑战。
新型(K)RAS抑制剂的分类
基于靶点选择性,新兴(K)RAS抑制剂可分为三类:
突变选择性抑制剂: 靶向特定(K)RAS突变(如G12C、G12D),同时保留所有野生型RAS亚型。
优势: 高特异性,潜在毒性较低。
挑战: 更易因旁路激活或继发突变产生耐药。
亚型选择性抑制剂: 靶向多种KRAS突变体(也称“泛KRAS抑制剂”)和野生型KRAS,同时最大限度减少对NRAS和HRAS的活性。
潜力: 可能覆盖>90%的KRAS突变癌症患者,同时通过降低对非KRAS亚型的活性来限制毒性,提供有利的平衡。
泛RAS抑制剂: 覆盖范围最广,靶向所有RAS亚型中的突变体和野生型。
优势: 可能提供更持久的靶点抑制和更广泛的疗效。
风险: 由于抑制全野生型RAS,对正常组织的靶上毒性风险更高。
功能与结合模式分类
基于结合模式:
靶向开关II口袋的抑制剂。
形成三元复合物的药物。
基于抑制状态:
可抑制GTP结合的激活态(ON state)的抑制剂(部分新一代药物具备此能力)。
其他策略:
除传统小分子抑制剂外,靶向蛋白降解(如PROTACs)等新方法正在研究中,旨在从细胞内清除突变KRAS蛋白。
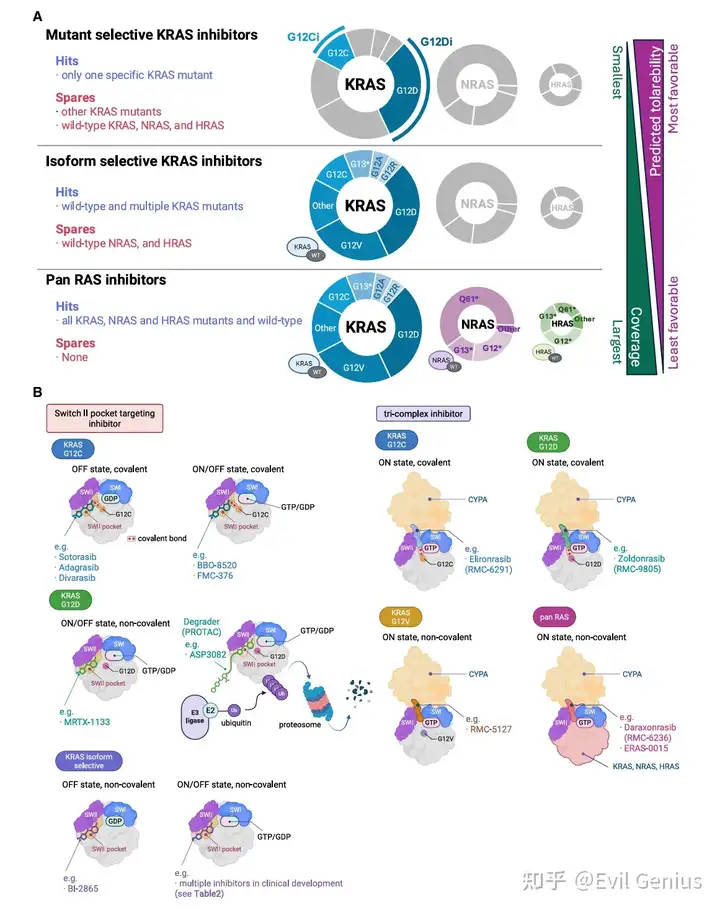
新兴(K)RAS抑制剂(突变选择性及更广谱抑制剂)总结
突变选择性抑制剂:针对特定KRAS突变
KRAS G12C激活态(ON State)抑制剂:
设计原理: 直接靶向更具挑战性的GTP结合的激活态,可克服对传统失活态(OFF State)抑制剂的耐药。
代表性药物:
Elironrasib (RMC-6291): 首创的共价三元复合物激活态抑制剂。通过细胞内伴侣蛋白亲环蛋白A(CYPA)形成稳定三元复合物,选择性共价抑制激活态KRAS G12C。临床前显示可克服开关II口袋耐药突变,初步临床试验在经治NSCLC中显示出疗效(ORR 50%),已获FDA突破性疗法认定。
BBO-8520 & FMC-376: 首创的共价双态抑制剂,可同时结合并抑制KRAS G12C的激活态和失活态。临床前显示对现有抑制剂耐药的模型(如KRAS扩增、RTK过表达)有效,已进入早期临床试验。
KRAS G12D选择性抑制剂:
挑战与策略: 缺少可共价结合的半胱氨酸,早期集中于开发非共价抑制剂。
代表性药物:
MRTX1133: 首个强效、选择性非共价KRAS G12D抑制剂,可结合并抑制其GDP和GTP两种状态。临床前有效,但因其药代动力学问题,临床开发已终止。
Zoldonrasib (RMC-9805): 首创的共价三元复合物激活态抑制剂,利用反应性氮丙啶弹头选择性共价结合KRAS G12D的Asp12。临床前及早期临床试验在NSCLC(ORR 61%)和PDAC(ORR 30%)中显示出积极疗效和良好安全性。
GFH375、HRS-4642、INCB161734: 其他双态KRAS G12D抑制剂,在早期临床试验中对NSCLC和PDAC也显示出有前景的初步疗效。
ASP3082: 首个靶向KRAS G12D的PROTAC分子,通过诱导蛋白降解而非抑制发挥作用,早期临床数据显示在NSCLC中ORR为38%。
针对其他突变(G12V, Q61H, G13C等)的抑制剂:
药物开发相对有限。针对第二常见突变KRAS G12V,RMC-5127成为首个报道的突变选择性非共价抑制剂,通过三元复合物机制靶向激活态。其他针对Q61H、G13C、G12S等的抑制剂尚在临床前阶段。
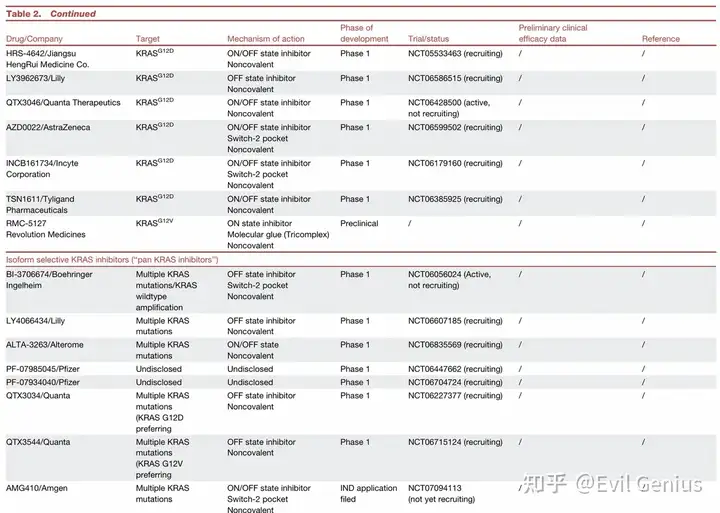
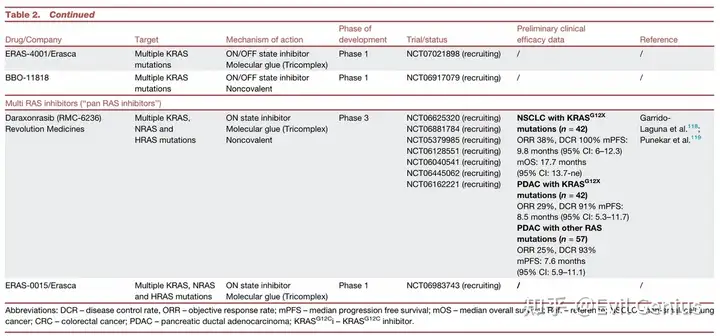
多RAS抑制剂(“泛RAS抑制剂”)
设计目标: 靶向所有RAS亚型(KRAS, NRAS, HRAS)的突变体和野生型,提供最广谱的覆盖,并可能预防野生型RAS介导的适应性耐药。
毒性担忧: 理论上有较高的靶上毒性风险,因为完全抑制RAS在正常组织中可能有害。
代表性药物:Daraxonrasib (RMC-6236)
机制: 首创的多RAS激活态抑制剂,通过CYPA依赖的“分子胶”机制形成三元复合物,干扰RAS-效应蛋白结合,并可刺激GTP水解。
特点: 临床前显示广谱抗肿瘤活性,能克服对G12C抑制剂的多种耐药机制(如二次突变、RTK改变)。治疗剂量下在小鼠中耐受良好。
临床数据: 在经治的晚期NSCLC和PDAC(含多种KRAS突变)中显示出有前景的疗效(PDAC: ORR 29%, mPFS 8.5个月; NSCLC: ORR 38%, mPFS 9.8个月)和可控安全性,已获FDA突破性疗法认定。III期试验正在进行中。
挑战: 已观察到靶向毒性(如痤疮样皮疹、胃肠道反应),且初步ORR低于最先进的突变选择性抑制剂。耐药机制(如KRAS扩增)也已出现。
亚型选择性KRAS抑制剂(“泛KRAS抑制剂”)
设计目标: 选择性靶向多种KRAS突变体及野生型KRAS,但不靶向NRAS或HRAS,旨在覆盖约90%的RAS突变,同时可能提供比泛RAS抑制剂更优的毒性谱。
代表性药物:BI-2865 / BI-3706674
机制: 非共价泛KRAS抑制剂,高选择性结合KRAS的失活态。
特点: 临床前对多种KRAS突变(G12C/D/V等)和扩增模型有效,尤其对野生型KRAS扩增的肿瘤细胞最敏感。
局限性: 缺乏对激活态的活性,因此在GTP水解严重受损(如G12R、Q61X突变)的肿瘤模型中效果有限。
未来方向: 下一代亚型选择性抑制剂正致力于开发能同时结合激活态和失活态的药物,以扩大治疗范围。
这篇文章既涉及癌症治疗领域KRAS抑制剂的相关内容,还点出多组学的发展趋势,更难能可贵的是坦诚分享了普通人做科研的现实困境,比如生存压力下进课题组或医院的不易,观点实在接地气。感谢作者的真诚分享,期待后续能看到更多实用内容。
这篇内容兼顾了专业领域和现实问题,既提到癌症治疗的KRAS抑制剂新格局与多组学发展趋势,又坦诚聊到普通出身者做科研的生存困境,没有空喊口号,还呼吁盗版商参与相关教程制作,内容很接地气,感谢分享,期待更多实在的科研相关内容。